
ADMET Predictions
Comprehensive prediction of absorption, distribution, metabolism, excretion, and toxicity using deep learning.
- AI-assisted analysis tools
- Lipinski & Veber compliance
- Instant comprehensive reports
Accelerate drug discovery with cutting-edge AI. From ADMET analysis to virtual screening and automated molecule generation, MolForge transforms your R&D pipeline.
Explore Popular Drugs
Search PubChem's database of compounds — instant ADMET predictions, molecular properties, and AI-assisted analysis.
Build, visualize, and analyze molecules in 3D. ADMET predictions, docking, QSAR — one unified workspace.
Launch BuilderPatent, literature & competitive signals across 100M patents and 276M papers — mapped to your molecule.
Open IP RadarChemBERTa, Mol2Vec, GNN embeddings & RL generation over 92M+ compounds — state-of-the-art, out of the box.
Explore ModelsDrug-polymer compatibility engine. Hansen screening, PC-SAFT thermodynamics, Tg mixing — plus AI-generated novel copolymers for ASD formulation.
Open Polymer LabBulk-test up to 10 drugs against the full pharma-polymer library. Sadowski-style PC-SAFT, Hansen distance, synergy scoring — one color-coded heat map, CSV export.
Open Solubility MatrixEvery FDA/EMA/PMDA approved drug — ATC class, protein targets, indications, adverse-event signals, pharmacogenomics, combinations, regulatory history. Cross-linked to 92 M compounds.
Explore DrugsMolForge combines AI with advanced scientific methods to deliver comprehensive molecular analysis and drug design capabilities.
Comprehensive prediction of absorption, distribution, metabolism, excretion, and toxicity using deep learning.

Screen massive compound libraries with AutoDock Vina and advanced scoring functions.

Design novel molecules using RL, VAE, and GANs optimized for drug-likeness.
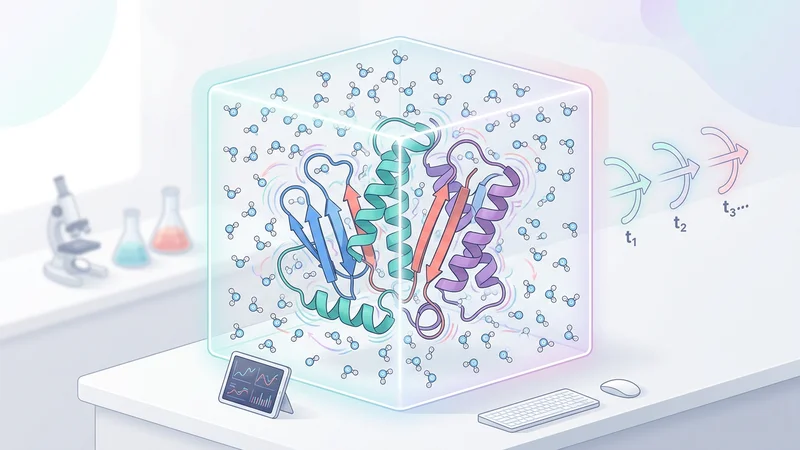
GROMACS integration for protein-ligand stability analysis and binding validation.

Comprehensive clustering, similarity analysis, and interactive 3D visualization.

Direct access to millions of compounds from the PubChem database.

Navigate interactive 2D and 3D maps of chemical neighborhoods with UMAP-projected molecular fingerprint space.
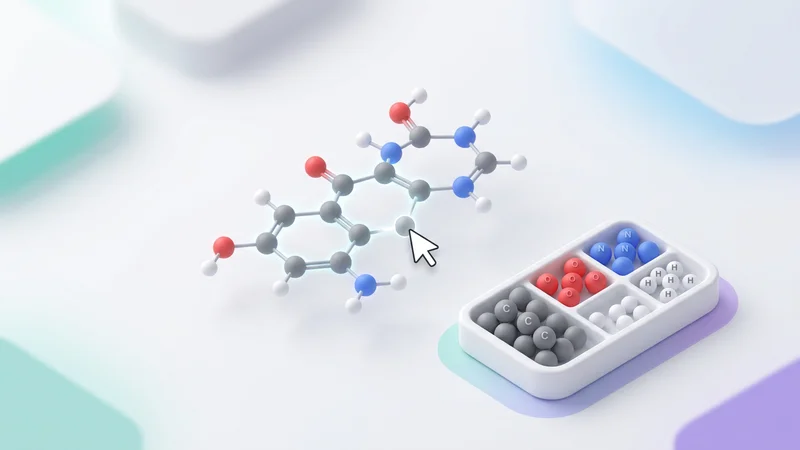
Design molecules atom-by-atom in an interactive 3D editor powered by Babylon.js with real-time property feedback.

Browse curated therapeutic targets across major disease areas and launch AI-assisted discovery programs.

Generate 12-section detailed molecular analysis PDFs covering properties, drug-likeness, bioactivity, and more.
Two AI radars working in parallel. Scan patents for freedom-to-operate. Scan literature for bioactivity. Land a defensible program in seconds.
Patent landscape & white-space map
Categorize claims, detect unpatented gaps, assess freedom-to-operate. 120M+ patents across USPTO, EPO, WIPO.
Scientific literature & bioactivity map
Parallel fan-out to OpenAlex, PubMed, Semantic Scholar. Extract IC50/Ki/EC50. Rank papers by citation influence.
No credit card required
Whether you're a student, researcher, or pharma executive — MolForge adapts to your workflow.
From target discovery to clinical trials, revolutionize every stage of pharmaceutical development with measurable results.

Leverage deep learning models to identify novel drug targets from biological networks and published literature.

Screen large compound libraries rapidly with GPU-accelerated docking and multi-objective filtering.

Generate novel drug candidates using reinforcement learning and generative models, optimized for drug-likeness.
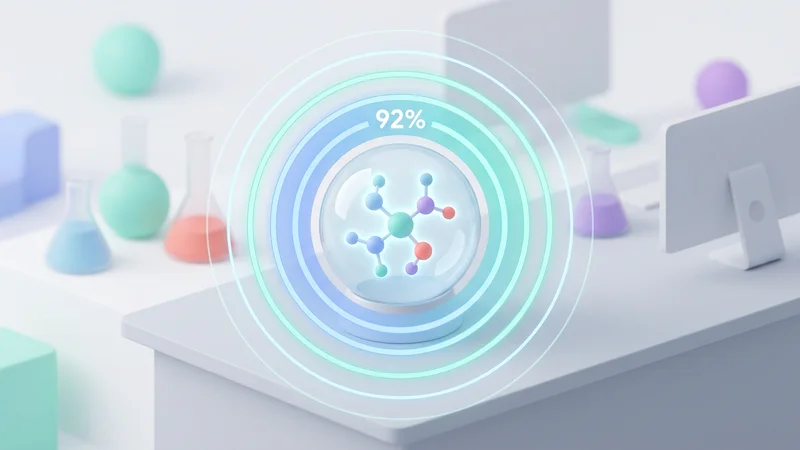
Predict absorption, toxicity, and metabolism early in the pipeline with ML models trained on curated datasets.
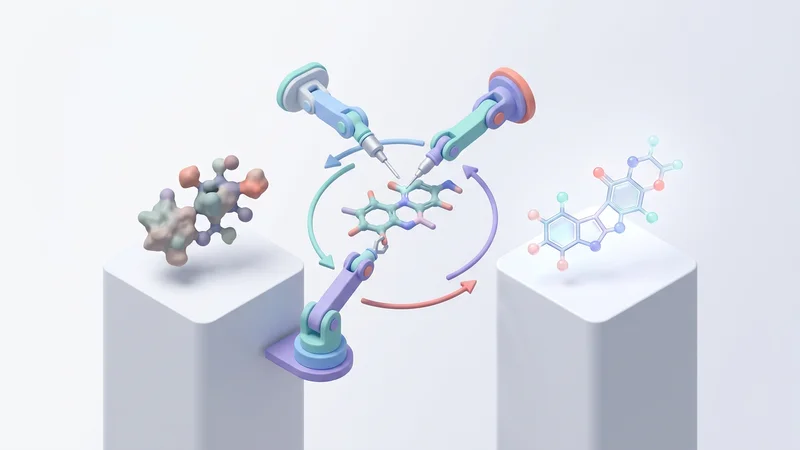
Transform hits into clinical candidates faster with multi-objective Bayesian optimization and SAR analysis.
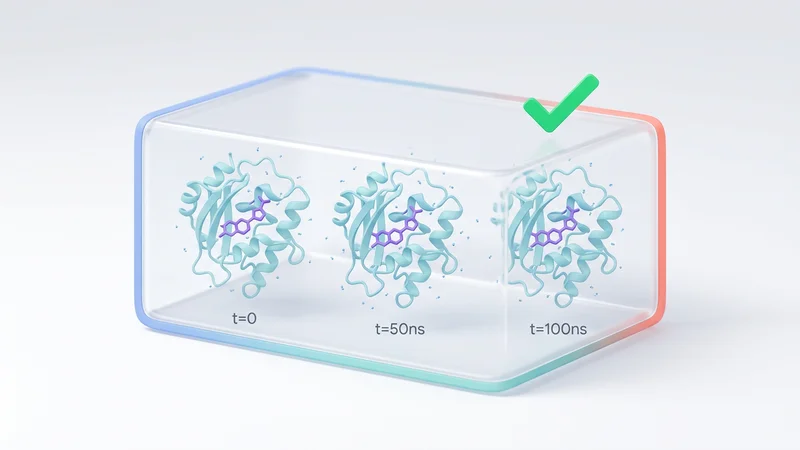
Run molecular dynamics simulations with automated GROMACS workflows and binding stability analysis.

Unify data from PubChem, ChEMBL, and your own sources into one searchable, AI-ready platform.
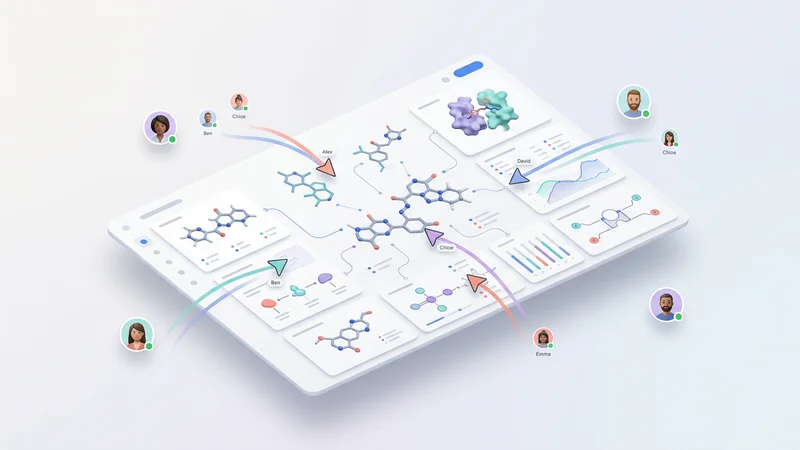
Streamline team collaboration with shared molecular projects, annotations, and role-based access.
Start free. Scale as you grow. No hidden fees, no surprises.
See all plans including Academic ($29/mo), Enterprise & API credits
Integrate molecular intelligence into your pipeline with our REST API. Property predictions, docking, QSAR, and more — all via simple HTTP requests.
import requests
response = requests.post(
"https://api.molforge.ai/v1/molecules/properties",
headers={"X-API-Key": "mf_your_key"},
json={"smiles": "CC(=O)Oc1ccccc1C(=O)O"}
)
print(response.json())Start using MolForge today and bring AI-powered molecular intelligence to your research pipeline.



